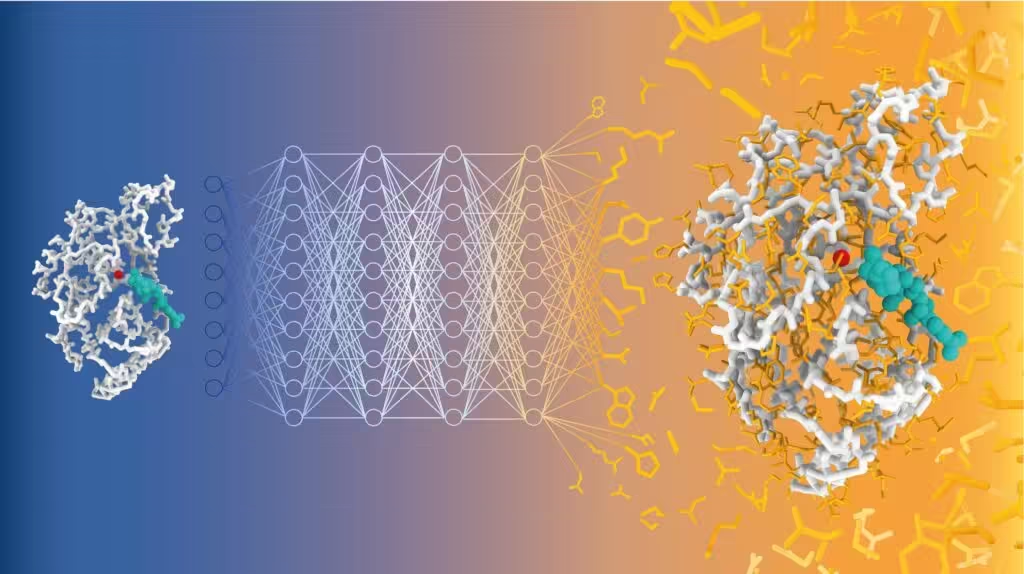
Illustration of sequence prediction with CARBonAra. The geometric transformer samples the sequence space of the TEM-1 beta-lactamase enzyme (in gray) complexed with a natural substrate (in cyan) to generate new, well-folded, and active enzymes. Credit: Alexandra Banbanaste (EPFL).
By Nik Papageorgiou
Designing proteins to perform specific functions requires a deep understanding and manipulation of their sequences and structures. This is essential for developing targeted treatments for diseases and creating enzymes for industrial applications.
One of the major challenges in protein engineering is designing proteins from scratch (de novo) to tailor their properties for specific tasks. This has significant implications for biology, medicine, and materials science. For instance, modified proteins can precisely target diseases, offering a competitive alternative to traditional small-molecule drugs.
Moreover, custom-designed enzymes, which act as natural catalysts, can facilitate rare or non-existent reactions in nature. This capability is particularly valuable in the pharmaceutical industry for synthesizing complex drug molecules and in environmental technology for more effectively breaking down pollutants or plastics.
A team of scientists led by Matteo Dal Peraro at EPFL has developed CARBonAra (Context-aware Amino acid Recovery from Backbone Atoms and heteroatoms), an AI-driven model that predicts protein sequences while considering constraints imposed by different molecular environments. CARBonAra is trained on a dataset of approximately 370,000 subunits, with an additional 100,000 for validation and 70,000 for testing, from the Protein Data Bank (PDB).
CARBonAra builds on the Protein Structure Transformer (PeSTo) framework—also developed by Lucien Krapp in Dal Peraro’s group. It uses geometric transformers, which are deep learning models that process spatial relationships between points, such as atomic coordinates, to learn and predict complex structures.
CARBonAra can predict amino acid sequences from backbone scaffolds, the structural frameworks of protein molecules. However, one of CARBonAra’s most remarkable features is its context-awareness, particularly in how it improves sequence recovery rates—the percentage of correctly predicted amino acids at each position of a protein sequence compared to a known reference sequence.
CARBonAra significantly improved recovery rates when including molecular « contexts, » such as protein interfaces with other proteins, nucleic acids, lipids, or ions. « The model is trained with all kinds of molecules and relies solely on atomic coordinates, allowing it to handle more than just proteins, » explains Dal Peraro. This feature enhances the model’s predictive power and applicability in complex, real-world biological systems.
The model performs well not only in synthetic benchmarks but has also been experimentally validated. Researchers used CARBonAra to design new variants of the TEM-1 β-lactamase enzyme, involved in antimicrobial resistance development. Some of the predicted sequences, differing by about 50% from the wild-type sequence, were correctly folded and retained some catalytic activity at high temperatures, where the wild-type enzyme is inactive.
The flexibility and precision of CARBonAra could pave the way for new avenues in protein engineering. Its ability to account for complex molecular environments could make it a valuable tool for designing proteins with specific functions, enhancing future drug discovery campaigns. Additionally, CARBonAra’s success in enzyme engineering demonstrates its potential for industrial applications and scientific research.
Read the full paper
Contextual geometric deep learning for protein sequence design, Lucien F. Krapp, Fernando A. Meireles, Luciano A. Abriata, Jean Devillard, Sarah Vacle, Maria J. Marcaida & Matteo Dal Peraro, Nature Communications (2024).

EPFL


